发病机制
发病机制
发病机制:
1.病理学 嗜铬细胞瘤来源于交感神经系统的嗜铬组织,分为散发型和家族型两大类。散发型嗜铬细胞瘤常为单个,80%~85%的肿瘤位于肾上腺内,右侧略多于左侧,少部分肿瘤位于肾上腺以外的嗜铬组织。家族型嗜铬细胞瘤常为多发性,也多位于肾上腺内,可累及双侧肾上腺,肾上腺外少见。在儿童患者中,肾上腺外和双侧肾上腺的嗜铬细胞瘤的发病率较高。肾上腺内的嗜铬细胞瘤直径常小于10cm。多为3~5cm,平均重量10g左右,大的肿瘤偶尔可超过1000g。肿瘤多为圆形或椭圆形,极少数为哑铃型;瘤体切面为灰色或棕褐色,或杂色相间,常有出血、坏死,囊性变或钙化,光镜下可见肿瘤由较大的,多角形的嗜铬细胞组成,在电子显微镜下可见细胞核周围有密集的富含肾上腺素和去甲肾上腺素的嗜铬颗粒。恶性嗜铬细胞瘤的直径较良性肿瘤大,在形态学上二者无明显差异,恶性者可有包膜的浸润,血管内可有瘤栓形成,但单凭显微镜所见很难鉴别,主要是观察其有无局部浸润和远处转移。转移的主要部位常为肝脏,骨骼,淋巴结和肺部。家族性嗜铬细胞瘤常为双侧多小结,多中心性病变,其恶性的发生率和复发率较散发型嗜铬细胞瘤高。
肾上腺外嗜铬细胞瘤(或称副神经节瘤)占散发型嗜铬细胞瘤的15%~20%,肾上腺外的肿瘤直径常小于5cm,重量在20~40g之间。肿瘤可在交感神经节内或节外,与肾上腺外嗜铬组织的解剖分布一致;大部分在腹部,可位于腹膜后腹主动脉前、左右腰椎旁间隙、肠系膜下动脉开口处、主动脉旁的嗜铬体(Zuckerkandl器),还可见于颈动脉体、颈静脉窦、肾上极、肾门、肝门、肝及下腔静脉之间、腹腔神经丛、近胰头处、髂窝或近髂窝血管处、卵巢内、膀胱内、直肠后等处;胸部的肿瘤常位于纵隔后交感神经干上,也可位于心包或心脏;马尾及其他部位的肿瘤罕见。约20%肾上腺外嗜铬细胞瘤是多发的。肾上腺外嗜铬细胞瘤恶性的发生率较大,表现为肿瘤切除后的复发和远处转移。肾上腺外嗜铬细胞瘤有多发、多病灶特点,要注意仔细查找,以防遗漏。
和其他内分泌腺肿瘤一样,肾上腺髓质肿瘤的病理诊断不能单靠形态表现,除激素测定和临床表现外,必须重视肿瘤细胞的生物学行为(激素合成、分泌和浸润能力)的评价。
在激素合成和分泌能力方面,用免疫组化方法可从瘤细胞中鉴定出如下激素:肾上腺素、去甲肾上腺素、多巴胺、血清素、乙酰胆碱、脑啡肽、CGRP、CRH、VIP、PACAP、ANP、AM、SS、神经肽Y、P物质、甘丙素等。一般肾上腺髓质的嗜铬细胞瘤的多激素分泌特点较肾上腺外者明显。
在遗传方面,散发性嗜铬细胞瘤的遗传标志不明,而家族性者(如MEN 2A型)多有明显的基因缺陷。最近的研究显示,嗜铬细胞瘤与副神经节瘤具有共同的染色体缺陷,用基因组比较杂交法发现两者的拷贝数变化很相似,两种肿瘤都存在1 cen-p3l(82%)及11q22-25(41%)等的丢失及其他改变。在组织病理形态学方面,单纯的细胞形态提供的诊断依据,特别是鉴别良恶性的依据是有限的,必须用免疫组织化学来协助鉴别。肿瘤细胞呈铬粒素、Leu7、S-100蛋白阳性反应仅说明其为神经外胚胎层来源,不能鉴别其良恶性。有时在细胞的生长、浸润行为模棱两可、确诊有困难时,可借助流式细胞仪诊断。如仍困难,则需依赖于临床的长期追踪观察。
本病的一般组织病理学诊断原则和方法可参照全美病理医师学院癌症委员会公布的诊断草案进行。
肾上腺髓质增生主要指嗜铬细胞的数目增多,按肾上腺髓质/皮质厚度比值计,如>1∶10认为可能有髓质增生。
肾上腺髓质增生可为单纯性或伴有MEN-Ⅱ.单纯性
肾上腺髓质增生大部分表现为双侧
肾上腺髓质增生,少数为单侧增生。有报道维生素D
3通过其促进有丝分裂的作用而使肾上腺髓质嗜铬细胞数量增加。另外,21-羟化酶缺陷者除有肾上腺皮质增生外,同时有肾上腺髓质功能减退和髓质增生。
肾上腺髓质增生的临床表现与嗜铬细胞瘤相似,有阵发性高血压和发作性高血压危象,血、尿儿茶酚胺及其代谢产物水平均可增高,但B超、CT及MRI不能发现肾上腺肿块,
131Ⅰ-MIBG可以表现为双侧或一侧(增生侧)肾上腺髓质摄取MIBG的量增多,确诊依靠病理学检查,病理改变为多发性结节性增生或弥漫性增生。手术治疗后血压可恢复正常。
一些免疫组化指标可用来判断肿瘤细胞的生物学行为。例如,单克隆抗体MIBl阳性细胞率在良恶性嗜铬细胞瘤中的差别很大,肾上腺的良性肿瘤细胞的MIBl阳性率低(0.81%)、恶性时高(3.30%);在肾上腺外,这种差别更明显(0.44%vs5.1%),故当MIB阳性细胞率>2%时,要高度疑为恶性嗜铬细胞瘤。
2.生化改变
(1)儿茶酚胺的合成、储存和释放:在嗜铬细胞瘤瘤细胞内的儿茶酚胺的合成和释放与正常肾上腺髓质中的嗜铬细胞不同,但嗜铬细胞瘤细胞中的嗜铬颗粒在形态和生理功能上与正常肾上腺髓质内的嗜铬颗粒完全一致。嗜铬颗粒内富含肾上腺素和去甲肾上腺素,但二者比例在不同的嗜铬颗粒内并不相同,由于肾上腺素(E)合成时必须有高浓度的糖皮质激素存在,故除肾上腺内及主动脉旁的嗜铬体内的肿瘤细胞产生较多的肾上腺素外,其他部位的肿瘤细胞一般仅能合成去甲肾上腺素(NE),此一特点对肿瘤的定位诊断有一定的帮助。可能是由于酪氨酸羟化酶的反馈抑制受到损害,儿茶酚胺的合成调节有所改变,肿瘤细胞合成儿茶酚胺的水平或多或少地要较正常的嗜铬细胞高。而且嗜铬细胞瘤不像正常的。肾上腺髓质一样受神经支配,儿茶酚胺的释放与神经冲动不一致,肿瘤的血流变化、直接加压、化学和药物刺激、血管紧张素-2的增加等均可引起肿瘤细胞组织中的儿茶酚胺释放,但其机制并不十分清楚。
(2)儿茶酚胺的排泄:不同于正常肾上腺髓质中的嗜铬颗粒(约85%为肾上腺素),大部分嗜铬细胞瘤中的嗜铬颗粒所含的NE较肾上腺素多,因此大部分病人尿中以去甲肾上腺素占优势。偶可全部是肾上腺素,从而临床上表现为β受体兴奋为主的症群如心动过速和高代谢状态,然而除非分别测定尿中的肾上腺素和去甲肾上腺素,大部分病人不可能从临床特征上来推断所排泄儿茶酚胺的种类。因其临床表现不典型,分泌、排泄肾上腺素的嗜铬细胞瘤诊断较为困难。肿瘤细胞仅合成和分泌肾上腺素(E)的机制未明。苯乙醇胺-N-甲基转移酶(PNMT)是催化NE转换为E的惟一限速酶,此类肿瘤细胞表达PNMT量大,与其他类型的嗜铬细胞瘤比较,PNMT仅在分泌E的肿瘤细胞中表达,并与17α-羟化酶及其受体蛋白一同表达。这提示,控制肾上腺素生成量的PNMT在肾上腺素分泌性嗜铬细胞瘤细胞呈过度表达,而过度表达的原因是皮质醇和Egr-1含量丰富。
家族型嗜铬细胞瘤患者肿瘤细胞内含有大量的儿茶酚胺,但血中和尿中的儿茶酚胺和儿茶酚胺代谢产物水平增加很少,特别是在多发性内分泌腺瘤综合征时,血浆中和尿中的肾上腺素增多仅仅是生化上的改变,一般不引起临床症状,其机制尚不清楚,此时诊断往往非常困难。
肾上腺外嗜铬细胞瘤除Zuckerkandl器的嗜铬细胞瘤外典型的仅分泌去甲。肾上腺素,但有报道胸腔内的嗜铬细胞瘤也可产生肾上腺素。在嗜铬细胞瘤早期,多巴胺和多巴胺代谢产物如高香草酸(HVA)的排泄常正常,如果尿液中多巴胺和HVA的排泄增加,多提示恶性的可能性较大。
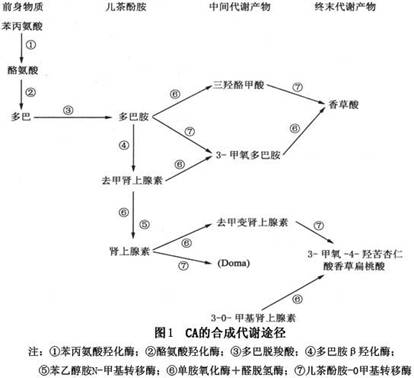
(3)肿瘤大小与儿茶酚胺水平:肿瘤的大小与游离的儿茶酚胺转化为儿茶酚胺代谢产物的比例有关。小的嗜铬细胞瘤,肿瘤内的儿茶酚胺的浓度低,但其排出多,故尿中VMA与CA的比例低;反之大的嗜铬细胞瘤,肿瘤内儿茶酚胺浓度高,但排出少,尿中VMA与儿茶酚胺的比例高。因为小肿瘤排出率高,因此分泌未代谢的儿茶酚胺,具有生物学活性并可产生临床表现,这类肿瘤往往在很小时即可诊断。相反,对于能储存较多的儿茶酚胺的肿瘤,在肿瘤内儿茶酚胺即可转化为其代谢产物,分泌有生物活性的儿茶酚胺少,因此在出现临床症状之前,肿瘤就已经较大。了解CA的合成及代谢过程将帮助我们对CA的生化来源,物质组成及代谢产物有明确的认识。其CA的合成代谢途径(图1)。
CA在体内是通过刺激受体而发挥作用的,其相关的肾上腺素能受体分为α、β及DA受体(DAC),各自又分为α1α2、β1β2及DAC1 DAC2受体。各种受体受刺激时的生理反应(表2)。
(4)肿瘤产生的其他物质:嗜铬细胞瘤除能合成肾上腺素和NE外,也能合成或分泌一些肽类物质,并且这些肽类在循环中的水平也可增高,其中包括促肾上腺皮质激素(ACTH)、促肾上腺皮质激素释放激素(CRH)、生长激素释放激素(GHRH)、降钙素基因相关肽(CGRP)、甲状旁腺素相关肽(PTHrP)、心钠素(ANP)、舒血管肠肽(VIP)、神经肽Y物质(NPY)、生长抑素、红细胞生成素及肾上腺髓质素(AM)、α-MSH等,这些肽类激素合成和分泌增多的机制不明,似乎不是由于神经刺激所致,可能与嗜铬颗粒分泌通道的反射性活动增加有关。这些肽类激素对临床表现有何影响也不十分清楚,但有些肽类可以引起特殊的内分泌综合征,如FTHrP分泌可引起继发性的
高钙血症,红细胞生成素分泌增多导致
继发性红细胞增多症。
3.家族型嗜铬细胞瘤及其相关疾病
(1)多发性内分泌腺瘤综合征:在多发性内分泌腺瘤Ⅰ型(MEN-1,Wermer综合征)中,嗜铬细胞瘤不常见。
多发性内分泌腺瘤Ⅱ型(MEN-2A,Sipple综合征)则包括嗜铬细胞瘤、甲状腺髓样癌和甲状旁腺瘤;约40%~50%的MEN-2A个体可发生嗜铬细胞瘤,其来源于
肾上腺髓质增生,常为多发性,双侧性的,肿瘤周围可有增生改变[弥漫性和(或)结节性增生],肾上腺外嗜铬细胞瘤罕见;肿瘤分泌的激素主要是肾上腺素,故早期临床症状可不典型,仅有血液或尿生化的改变。多发性内分泌腺瘤Ⅲ型(MEN-2B)由神经节神经瘤表现型(类马凡体型,多发性黏膜神经瘤)、甲状腺髓样癌和嗜铬细胞瘤组成,嗜铬细胞瘤的发病率为40%~50%。
(2)von Hippel-Lindau综合征(VHL综合征):von Hippel-Lindau综合征是一种常染色体显性遗传病,由视网膜
血管瘤、中枢神经血管网状细胞瘤、肾癌、肾脏和胰腺囊肿及多发囊腺瘤组成。嗜铬细胞瘤的发生率占10%~20%,常为多发性的,在不同家族中嗜铬细胞瘤的发病率不同,在某些家族中可高达90%。且其发生往往较早。
本综合征的病因和发病机制已基本查清,染色体3p25区含有肿瘤抑制基因(tumol supprssor gene,TSG),由于TSG的突变或缺失可导致血管性肿瘤(良性)、囊肿(肝、肾、胰腺等)和囊腺瘤的形成。约70%的病人可伴有肾透明细胞癌,嗜铬细胞瘤的外显率差别很大。但近年来有较多报道指出,本征易合并内淋巴囊肿瘤(endolymphtic sac tumors)是耳鸣和
耳聋的重要病因之一。在临床上,遇到家族性视网膜、脑组织的血管细胞瘤或多发性胰腺囊肿时要想到VHL综合征可能,但单凭附睾或肾囊肿不能诊断为VHL。对无家族史者,必须在具备两个或两个以上的视网膜和(或)脑组织血管网状细胞瘤时,或在具备一个血管网状细胞瘤伴一个内脏肿瘤时,才可作出VHL的临床诊断。确诊有赖于TSG基因突变的分子生物学检查或证明有3p 25区的缺失存在。凡家族成员都必须作DNA或3P 25区缺失的筛查试验,阳性携带者必须接受严密的追踪观察。
TSG基因(又称VHL基因)含3个外显子,编码两种mRNA,约20%的病人用Southerm分析可查出有生殖系突变(累及所有细胞),27%有无义突变或移码(frameshift)突变,VHL家族成员中阳性检出率约80%,家族中患嗜铬细胞瘤者(VHL2型)约占7%~20%,多数VHL 2型家族中的VHL,基因为无义突变,而VHL 1型家族(不患嗜铬细胞瘤)的VHL基因为完全缺乏或部分缺乏(因终止密码子提前出现所致)。VHL的临床不均一性来源于基因缺陷和外显率的不均一性,有时,还与肿瘤细胞存在嵌合染色体有关。
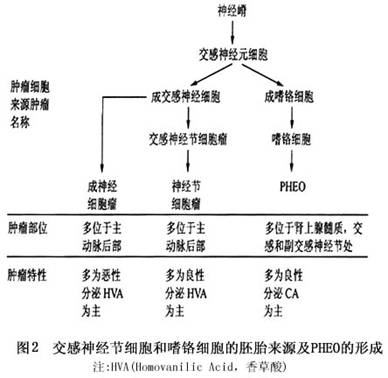
肾上腺髓质和交感神经系统共同起源于胚胎期的交感神经元细胞,经分化演变而成为交感神经节细胞及嗜铬细胞,这些细胞的异常分化而形
成神经细胞瘤、神经节瘤及PHEO。其分化及演变过程(图2)。
(3)多发性神经纤维瘤和其他相关疾病:多发性神经纤维瘤有两种亚型(Ⅰ型和Ⅱ型),嗜铬细胞瘤只与Ⅰ型有关,其发病率在多发性神经纤维瘤的人群中高低不一,1%~50%。在Carney复合征中.常表现为有功能性的肾上腺外的副神经节瘤。另外在Sturge-Weber综合征中也常伴有嗜铬细胞瘤。
Yokoyma等用甲氧氯普胺(胃复安)试验作为可疑患者的激发试验,用于鉴别肾上腺肿块的性质。7例嗜铬细胞瘤病人中,3例阳性,3例阴性,1例假阴性,其敏感性和特异性与24h尿儿茶酚胺测定和MIBG相似,而特异性均为100%,阴性结果并非试验不敏感,而是疾病本身具有不均一的生物学特征所致。曾认为甲氧氯普胺(胃复安)可促进AVP(ADH)分泌,但Coiro等用20mg甲氧氯普胺(胃复安)1次静脉注射,未发现血浆AVP有任何变化。Hsu等发现,嗜铬细胞瘤(6/7)呈阳性反应,除血压升高外,同时观察试验前后的血浆和尿儿茶酚胺变化更有诊断意义,但试验前应停用干扰CA分泌的药物。
临床表现
临床表现:本病的临床表现个体差异甚大:从无症状和体征至突然发生
恶性高血压、心衰或
脑出血等。其常见症状和体征如下:
1.心血管系统
(1)高血压:为本症的主要和特征性表现,可呈间歇性或持续性发作。典型的阵发性发作常表现为血压突然升高,可达200~300/130~180mmHg,伴剧烈
头痛,全身大汗淋漓、心悸、心动过速、
心律失常,心前区和上腹部紧迫感、疼痛感、焦虑、恐惧或有濒死感、皮肤苍白、恶心、呕吐、
腹痛或胸痛、视力模糊、复视,严重者可致
急性左心衰竭或心脑血管意外。发作终止后,可出现面部及全身皮肤潮红、发热、
流涎、瞳孔缩小等迷走神经兴奋症状和尿量增多。阵发性发作可由情绪激动、体位改变、创伤、灌肠、大小便、腹部触诊、术前麻醉或某些药物(如组胺、胍乙啶、胰高糖素、多巴胺拮抗剂、安非他命、儿茶酚胺再摄取阻断剂和单胺氧化酶抑制剂等)促发。发作持续时间不一,短至数秒或长至数小时以上。发作频率不一,多者1天数次,少者数月1次。随病程进展发作渐频渐长,一般对常用的降压药效果不佳,但对α-肾上腺能受体拮抗剂、钙通道阻滞剂有效。若高血压同时伴上述交感神经过度兴奋、高代谢、
头痛、焦虑、烦躁、直立性
低血压或血压波动大,尤其发生于儿童或青年时,应高度怀疑为本病。少数患者(多为儿童或青年)可表现为病情发展迅速,呈急进性
恶性高血压,舒张压可高于130mmHg,眼底损害严重,短期内可出现
视神经萎缩以及失明,可发生氮质血症、心衰或高血压脑病。
(3)心脏:大量儿茶酚胺可致儿茶酚胺性心脏病,可出现
心律失常如期前收缩、阵发性心动过速、心室颤动。部分病例可致心肌退行性变、坏死、炎性改变等心肌损害,而发生心衰。长期、持续的高血压可致左心室肥厚、心脏扩大和心力衰竭。
2.代谢紊乱 高浓度的肾上腺素作用于中枢神经系统,尤其是交感神经系统而使耗氧量增加,基础代谢率增高可致发热、消瘦。肝糖原分解加速及胰岛素分泌受抑制而使糖耐量减退,肝糖异生增加。血糖升高及出现尿糖。大量儿茶酚胺又可加速脂肪分解,使血游离脂肪酸增高而致血脂异常。大量儿茶酚胺也可促使血钾进入细胞内及。肾素和醛固酮分泌增加,排钾过多,少数可出现
低钾血症。也可因肿瘤分泌甲状旁腺激素相关肽(PTHrP)而致
高钙血症。
3.其他表现 过多的儿茶酚胺使肠蠕动及张力减弱,故可致
便秘、肠扩张、胃肠壁内血管发生增殖性或闭塞性动脉内膜炎,致肠坏死、出血或穿孔;胆囊收缩减弱,Oddi括约肌张力增强,可致胆汁潴留、胆结石。病情严重而病程长者可致肾衰竭。膀胱内嗜铬细胞瘤患者排尿时,可诱发血压升高。在大量肾上腺素作用下血细胞发生重新分布,使外周血中白细胞增多,有时红细胞也可增多。此外,本病可为Ⅱ、Ⅲ型多发性内分泌腺瘤综合征(MEN)的一部分,可伴发甲状腺髓样癌、甲状旁腺腺瘤或增生、肾上腺腺瘤或增生。
治疗
治疗:嗜铬细胞瘤一旦确诊并定位,应及时切除肿瘤,否则有肿瘤突然分泌大量CA、引起高血压危象的潜在危险。
术前应采用α受体阻滞剂使血压下降,减轻心脏负荷,并使原来缩减的血管容量扩大,以保证手术的成功。
1.术前准备和药物治疗
(1)α-肾上腺素能受体阻断剂:
①酚妥拉明(phentolamine,Regitine):用于高血压的鉴别诊断(Regitine试验),治疗高血压危险发作或手术中控制血压,而不适于长期治疗。
②
酚苄明(phenoxybenzamine):常用于术前准备,术前7~10天,初始剂量10mg/d,口服,平均递增0.5~1.0mg/(kg・d),分为2次/d,直至血压接近正常,大多数患者约需40~80mg/d。服药过程中应严密监测卧、立位血压和心率的变化。
③哌唑嗪(prazosin)、特拉唑嗪(terazosin)、多沙唑嗪(doxazosin):均为选择性突触后α1肾上腺素能受体阻滞剂。应用时易致严重的直立性低血压,故应在睡前服用,尽量卧床。
④乌拉地尔(urapidil,压宁定):可阻断α1、α2受体,并可激活中枢5-羟色胺1A受体,降低延髓心血管调节中枢的交感反馈作用,故在降压的同时不增加心率。
(2)β肾上腺素能受体阻断剂 因使用α受体阻断剂后,β受体兴奋性增强而致心动过速、心收缩力增强、心肌耗氧量增加,应使用β受体阻滞剂改善症状,但不应在未使用α受体阻断剂的情况下单独使用β受体阻断剂,否则可能导致严重的肺水肿、心衰或诱发高血压危象等。
①普萘洛尔(心得安):初始剂量10mg,2~3次/d,可逐渐增加剂量,以达到控制心率的目的。
②阿替洛尔(氨酰心安):常用剂量25~50mg,2~3次/d,无明显负性心肌收缩作用。
③美托洛尔(美多心安):常用剂量50mg,2~3次/d。
④艾司洛尔(esmolol):静脉滴注,可迅速减慢心率。
(3)钙通道阻断剂(CCB)CCB可用于术前联合治疗,尤适用于伴冠心病或CA心肌病患者,或与α、β受体阻断剂合用进行长期降压治疗。常用硝苯地平(nifedipine),口服,10~30mg/d。
(4)血管紧张素转换酶抑制剂(ACEI) 如卡托普利(captopril),口服,12.5~25mg,3次/d。
(5)血管扩张剂:硝普钠(sodinm nitroprusside)是强有力的血管扩张剂,主要用于嗜铬细胞瘤患者的高血压危象发作或手术中血压持续升高者。用5%
葡萄糖液溶解和稀释,从小剂量开始,逐渐增强至50~200μg/min。严密监测血压,调整药物剂量,以防血压骤然下降,并监测氰化物的血药浓度。
(6)儿茶酚胺合成抑制剂:α-甲基对位酪氨酸(α-methyl paratyrosine)为酪氨酸羟化酶的竞争性抑制剂,阻断CA合成。口服初始剂量为250mg,6~8小时1次,根据血压及血、尿CA水平调整剂量,可逐渐增加。总剂量为1.5~4.0g/d。常见的副作用有嗜睡、抑郁、消化道症状、锥体外系症状如帕金森症候群等。减量或停药后上述症状可很快消失。
2.131Ⅰ-MIBG治疗 主要用于恶性及手术不能切除的嗜铬细胞瘤,常用剂量为100~250mCi。
3.嗜铬细胞瘤所致高血压危象的治疗 应首先抬高床头,立即静脉注射酚妥拉明1~5mg。密切观察血压,当血压降至160/100mmHg左右时,停止注射。继之,以10~15mg溶于5%
葡萄糖生理盐水500ml中,缓慢滴注。
4.术后处理 在肿瘤切除后,患者血压很快下降。如术后仍存在持续性高血压,可能是肿瘤未切除干净或已伴有原发性高血压或肾性高血压。儿茶酚胺在手术后7~10天即可恢复正常水平。因此在术后1周时要测定CA或其代谢物以明确肿瘤是否完全切除。
对于不能手术的患者或者恶性肿瘤扩散的患者,可以长期药物治疗。多数的肿瘤生长很慢。应用肾上腺素能受体阻滞剂以及a甲基酪氨酸长期治疗可有效抑制儿茶酚胺合成。
5.恶性嗜铬细胞瘤的治疗 恶性嗜铬细胞瘤可以在腹膜后复发或是转移到骨、肺、肝脏等处。复发有可能在第1次术后的数年或数十年后才发生,需要长期随诊观察。放疗虽效果不是很好,但对控制骨转移有好处。可以联合应用环磷酰胺、长春新碱、达卡巴嗪(甲氮咪胺)化疗,但成功的报道也不多。131Ⅰ-MIBG治疗也有报道。
6.家族性嗜铬细胞瘤的处理 家族性嗜铬细胞瘤通常是多发的或是累及双侧肾上腺,而且复发率高。其治疗还是一个难题。可供选择的方案有对小的、无功能的肿瘤进行随诊观察、肿瘤侧肾上腺切除、预防性双侧肾上腺切除等。在双侧肾上腺全切术后应注意长期皮质激素替代治疗。
7.妊娠期嗜铬细胞瘤的处理 孕期嗜铬细胞瘤较难处理。在未经任何准备的情况下经阴道自行分娩往往会给产妇及婴儿带来很大危害。肿瘤的定位适宜行M
RI检查,不会有副作用。一旦诊断明确,就应服用α受体阻滞剂控制症状。如果是在妊娠的早期及中期,如术前准备充分后应立即手术。术后不需要终止妊娠,但手术有可能增加流产的几率。如果诊断时已处于妊娠晚期,在胎儿足月时可以随嗜铬细胞瘤手术而行剖宫产。如胎儿尚未成熟,应继续服用药物,并进行严密的监护,直到适宜手术。但如果在监护过程中病情进展很快,手术不能拖延。尽管在孕期服用肾上腺素能受体阻滞剂是否影响胎儿的发育还不太明确,但临床上已应用于不少病例,没有出现明显的副作用。